From FASTQ to CNV calls in Julia
I’ve recently worked on Julia bindings for BWA, allowing to align sequencing reads (FASTA or FASTQ) to a reference genome directly from Julia. On top of this, updates to XAM.jl and BioRecordsProcessing.jl will make it possible to build end-to-end workflows in Julia, from raw reads to analysis results.
To illustrate this I will showcase online whole-genome sequencing (WGS) copy number variation (CNV) calling on a mouse kidney tumor sample.
Briefly, we need to do the following :
- Read a pair of FASTQ files
- Align each pair of reads to the reference genome
- Count the number of read pairs aligning to a given window in the reference genome
- Normalize the counts to produce a normalized (log2) coverage profile
- Use an HMM to segment the profile
- Visualize the results
This will result in the following animation, showing how the coverage profile and segmentation evolves as more reads get processed :
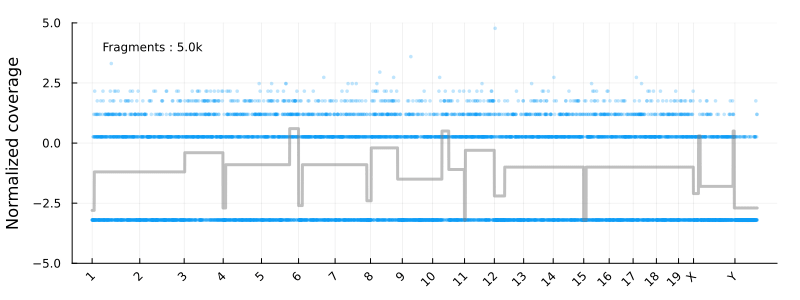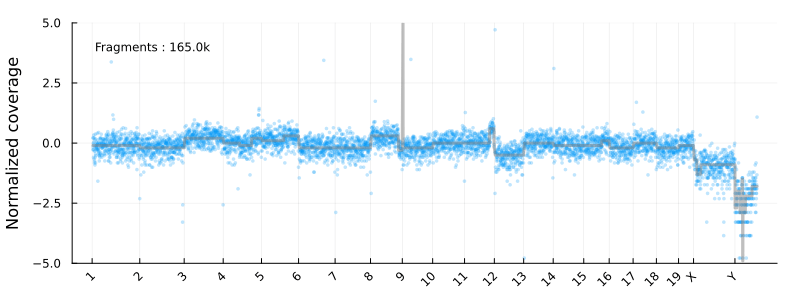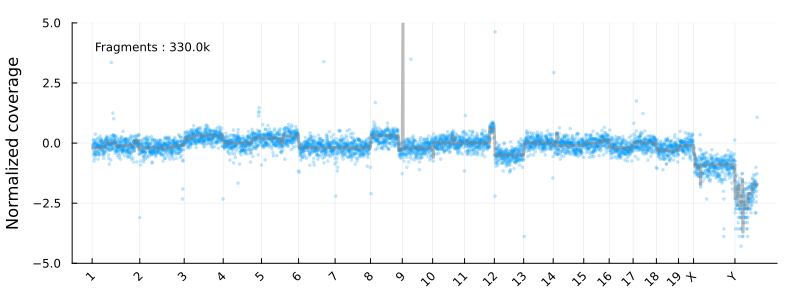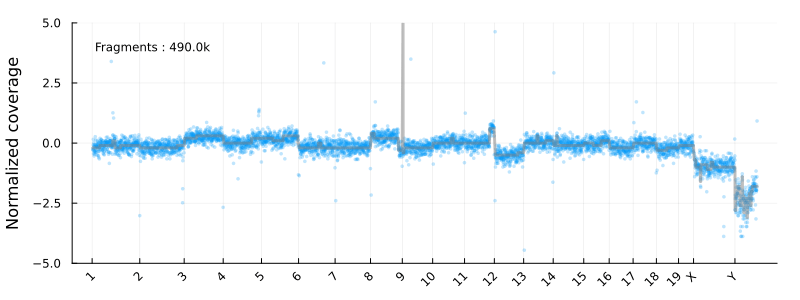
Disclaimer : This is not intended to be used as a serious CNV calling pipeline. I’m also using the development versions of the libraries listed above. See e.g. this article for a review of WGS CNV calling tools.
Aligning reads to the reference genome
First we need to get some data, finding publicly available WGS data from tumor is difficult, but after browsing I found this Mus musculus kidney tumor sample that has WGS data that can be downloaded from NCBI.
Next we’ll setup an aligner from BurrowsWheelerAligner.jl, loading the reference genome (which was already indexed with bwa index) :
using BurrowsWheelerAligner
const BWA = BurrowsWheelerAligner
aligner = BWA.Aligner(
"/tmp/mouse_genome/Mus_musculus.GRCm39.dna.primary_assembly.fa.gz";
paired = true, nthreads = 2
)
Given a pair of FASTQ reads, to obtain an alignment (as a XAM.SAM.Record) we simply do :
sam_r1, sam_r2 = BWA.align(aligner, (read1, read2))
julia>sam_r1
XAM.SAM.Record:
template name: SRR24288063.1
flags: 83
reference: 13
position: 51515489
mapping quality: 60
...
Typically we want to save these aligned reads to a BAM file. BWA internal alignment methods returns a C string
, these are parsed and wrapped in a SAM.Record, which then needs to be encoded
in the proper binary representation of the BAM format. This can simply done with XAM by passing a SAM.Record to the BAM.Record constructor : XAM.BAM.Record(sam_r1).
Building a pipeline
In order to read from our pair of FASTQ files and write to a BAM file we will use BioRecordsProcessing.jl,
which will do the file management for us. In BioRecordsProcessing a pipeline is built by providing a
source - which will read records and pass them to a user-defined processing function - and a sink that will
write the records to an output file or collect them in memory. For our case the pipeline will look like :
using BioRecordsProcessing, XAM, FASTX
file = "SRR24288063_1.fastq.gz"
header = BWA.header(aligner) # SAM.Header containing the chromosome names and lengths
p = Pipeline(
# Source
Reader(FASTQ, File(file; second_in_pair = x -> replace(x, "_1" => "_2"))),
# User-defined function
(reads...) -> begin
r1, r2 = BWA.align(aligner, reads)
return BAM.Record(r1; header=header), BAM.Record(r2; header=header)
end,
# Sink
Writer(
BAM, ".";
extension = ".bam", header = header, paired = false
),
)
run(p; max_records = 2) # align two fragments
Note we passed the header when converting the records to BAM, this is needed to get the proper chromsome names in the BAM file. We can check that our BAM file is indeed correct :
run(`samtools view SRR24288063_1.fastq.bam`)
# SRR24288063.1 83 13 51515489 60 144M7S = 51515464 -169 TTCTCTGAGCTTCA...
Counting read-pairs and coverage profile
Now that we can align our reads we want to count how many pairs fall in a given genomic interval. To do so, we define 500kb bins from chromsome 1 to chromosome Y, with the first bin of chromsome 2 following the last of chromosome 1 and so on until the end of Y :
Chr1 Chr2
-------------- -------------- ...
BIN1 BIN2 BIN3 BIN4 BIN5 BIN6
We define a function to_bin(chr, pos) taking a chromosome and genomic position to quickly
compute the index of the bin corresponding to the genomic position.
chromosomes = vcat(string.(1:19), "X", "Y")
# get chromosome length from header
chromosome_lengths = [
first(filter(h -> SAM.tag(h) == "SQ" && h["SN"] == chr, header.metainfo))["LN"] |> x->parse(Int,x)
for chr in chromosomes]
# define bins
binsize = 500_000
bins = 1:binsize:sum(chromosome_lengths)
offsets = Dict(zip(chromosomes, vcat(0, cumsum(chromosome_lengths)[1:end-1])))
to_bin(chr, pos) = ceil(Int, (pos + offsets[chr]) / binsize)
# initialize counts to zero
counts = zeros(Int, length(bins))
We can update our processing function with:
bin_idx = to_bin(SAM.refname(r1), SAM.position(r1))
counts[bin_idx] += 1
if mod(k, 5_000) == 0
update_plot(bins, count, offsets, anim; segmentation = nothing)
end
k += 1
Where update_plot is normalizing the counts and plot them using Plots.jl. The chromsomes are shown on the x-axis
and the y axis is the log2 normalized coverage. In these coordinates -1 and 1 corresponding to copy numbers 1 and 4 for diploid cells
at high tumor purity.
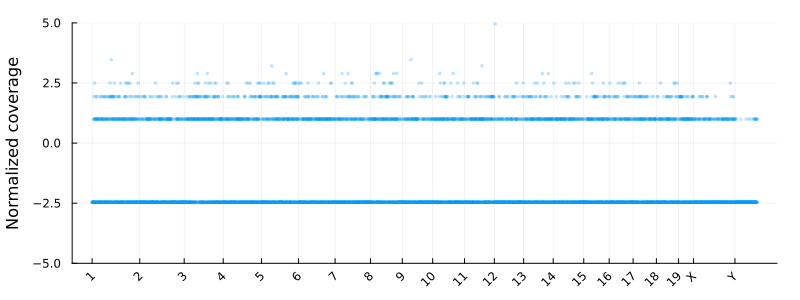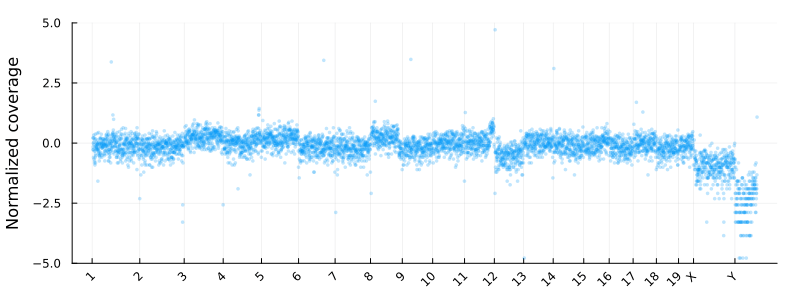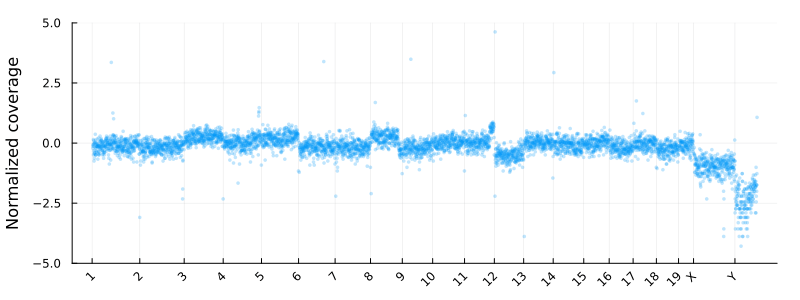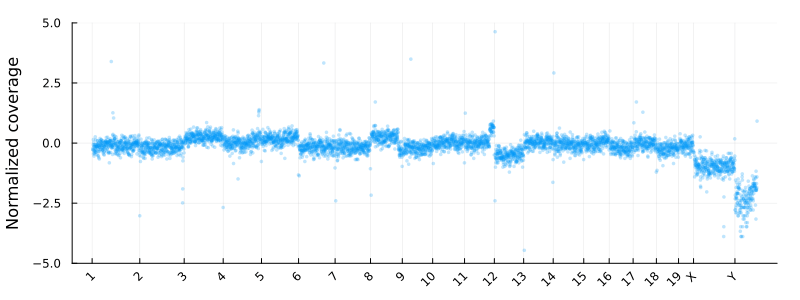
Segmentation using a HMM
Finally we’ll use a simple hidden Markov model to segment the counts. I won’t go into details
here, but shortly I’m using a fixed discretisation of the normalized coverage (from -5 to 5) for the hidden states,
and a Normal emission with a standard deviation that decreases with increasing number of reads. There’s a small, uniform probability
of transitioning from a hidden state to any other. Finally I use viterbi to get the most likely sequence of hidden states.
using HiddenMarkovModels, LinearAlgebra, Distributions
function segment_profile(counts)
bins_cov = -5:0.1:5
σ = 1/sqrt(mean(counts))
N = length(bins_cov)
p_transition = 1e-6
transitions = I(N)*(1-p_transition) .+ p_transition
transitions = transitions ./ sum(transitions, dims=1)
# mix-in some uniform distributio to help with outliers
get_emission(μ) = MixtureModel(
vcat(Normal(μ, σ), Uniform(-5,5)), [0.99, 0.01]
)
emissions = [get_emission(mu) for mu in bins_cov]
initial_state = ones(N)/N
hmm = HMM(initial_state, transitions, emissions)
obs_seq = log2.((counts .+ 0.1) ./ mean(counts))
obs_seq = round.(obs_seq, digits = 1)
obs_seq = clamp.(obs_seq, extrema(bins_cov)...)
most_likely_state_seq, L = viterbi(hmm, obs_seq)
seg = bins_cov[most_likely_state_seq]
seg
end
Producing the animation shown above :
Performance
Processing 10k read pairs with my pipeline takes about 30s, while simply aligning them with bwa mem
took about 7 seconds, or a 4x slowdown. Both methods used 2 threads. However, profiling my Julia code indicates
that most time is spent in BWA’s internal mem_process_seqs method. I’m calling this method for each read pair, it might
be better to gather a batch of reads and then pass them to the method.
Final code
using BioRecordsProcessing, XAM, FASTX, BurrowsWheelerAligner, Plots, Statistics
using HiddenMarkovModels, LinearAlgebra, Distributions
const BWA = BurrowsWheelerAligner
aligner = BWA.Aligner(
"/mnt/ssd_disk/mouse_genome/Mus_musculus.GRCm39.dna.primary_assembly.fa.gz";
paired = true, nthreads = 2
)
header = BWA.header(aligner)
chromosomes = vcat(string.(1:19), "X", "Y")
# get chromosome length from header
chromosome_lengths = [
first(filter(h -> SAM.tag(h) == "SQ" && h["SN"] == chr, header.metainfo))["LN"] |> x->parse(Int,x)
for chr in chromosomes]
# define bins
binsize = 500_000
bins = 1:binsize:sum(chromosome_lengths)
offsets = Dict(zip(chromosomes, vcat(0, cumsum(chromosome_lengths)[1:end-1])))
to_bin(chr, pos) = ceil(Int, (pos + offsets[chr]) / binsize)
# initialize counts to zero
counts = zeros(Int, length(bins))
function segment_profile(counts)
bins_cov = -5:0.1:5
σ = 1/sqrt(mean(counts))
N = length(bins_cov)
p_transition = 1e-6
transitions = I(N)*(1-p_transition) .+ p_transition
transitions = transitions ./ sum(transitions, dims=1)
get_emission(μ) = MixtureModel(
vcat(Normal(μ, σ), Uniform(-5,5)), [0.99, 0.01]
)
emissions = [get_emission(mu) for mu in bins_cov]
initial_state = ones(N)/N
hmm = HMM(initial_state, transitions, emissions)
obs_seq = log2.((counts .+ 0.1) ./ mean(counts))
obs_seq = round.(obs_seq, digits = 1)
obs_seq = clamp.(obs_seq, extrema(bins_cov)...)
most_likely_state_seq, L = viterbi(hmm, obs_seq)
seg = bins_cov[most_likely_state_seq]
seg
end
function update_plot(bins, counts, offsets, anim; segmentation = nothing)
p = scatter(
bins, log2.((counts .+ 0.1) ./ mean(counts)),
alpha = 0.25, markerstrokewidth = 0, markersize = 2,
size = (800,300), legend = false, margin = 15Plots.px,
xticks = (values(offsets), keys(offsets)),
xlim = extrema(bins),
ylim = (-5,5),
xrotation = 45,
ylabel = "Normalized coverage",
)
annotate!(bins[500], 4, text("Fragments : $(sum(counts)/1000)k", 8))
if !isnothing(segmentation)
sel = segmentation .!= -5
plot!(bins[sel], segmentation[sel], c="gray", alpha = 0.5, linewidth=3)
end
frame(anim)
display(p)
end
function align_fastq(aligner, file, bins, counts, max_records)
k = 1
anim = Animation()
p = Pipeline(
Reader(FASTQ, File(file; second_in_pair = x -> replace(x, "_1" => "_2"))),
(reads...) -> begin
r1, r2 = BWA.align(aligner, reads)
!SAM.hasrefname(r1) && return nothing
!SAM.hasrefname(r2) && return nothing
!haskey(offsets, SAM.refname(r1)) && return nothing
bin_idx = to_bin(SAM.refname(r1), SAM.position(r1))
counts[bin_idx] += 1
if mod(k, 5_000) == 0
segmentation = segment_profile(counts)
update_plot(bins, counts, offsets, anim; segmentation = segmentation)
end
k += 1
return BAM.Record(r1; header=header), BAM.Record(r2; header=header)
end,
Writer(
BAM, ".";
extension = ".bam", header = BWA.header(aligner), paired = false
),
)
out = run(p; max_records = max_records)
gif(anim, "$(file)_segmentation.gif")
out
end
file = "SRR23498664_1.fastq"
max_records = 500_000
@time align_fastq(aligner, file, bins, counts, max_records)